
کاهش مصرف اکسیژن توسط سلول های چربی، نقصهای متابولیکی را بهبود می بخشد
سطوح پایین اکسیژن نشانه ی رشد بافت چربی در چاقی است و می تواند به دیابت نوع 2 منجر شود. همراه با فقدان خون رسانی کافی، نیاز به افزایش اکسیژن در سلولهای چربی اکنون به عنوان کلید اصلی این وضعیت مضر پدیدار شده است.
یکی از دلایل اصلی دیابت نوع 2، چاقی است که در آن سلولهای چربی به سرعت از نظر اندازه و تعداد گسترش می یابند و همین رشد زیاد سبب می شود که نیاز به اکسیژن آنها فراتر از اکسیژن عرضه شده باشد. این کمبود اکسیژن، که به عنوان هیپوکسی شناخته می شود، منجر به افزایش تولید پروتئین ضد هیپوکسی HIF-1α می شود که به نوبه خود سبب التهاب بافت می شود و از پاسخ طبیعی سلول های چربی (آديپوسیت ها) به انسولین جلوگیری می کند. اغلب تصور می شود که هیپوکسی در بافت چربی در حال رشد، به دلیل مشکل عرضه ی اکسیژن اتفاق می افتد، زیرا عروق خونی توانایی لازم را برای رساندن اکسیژن به بافت وسیع چربی با همان سرعت اکسیژن رسانی به بافتهای اطراف را ندارند. دکترSeo و همکارانش در مقاله ی خود که در مجله ی Nature Metabolism منتشر شده است، بر مسیری تاکید کردند که توسط آن آديپوسیت ها با مصرف اکسیژن بیش از حد، در افزایش هیپوکسی در بافت چربی در حال رشد نقش دارند. در این مسیر سلولی، فعالیت آنزیم آدنین نوکلئوتید ترانسلوکیز 2 (ANT2) در اندامکهای تولید انرژی یعنی میتوکندریها افزایش می یابد.
در طی تنفس طبیعی میتوکندری، الکترون ها بین یک سری از مولکول ها منتقل می شوند و این انتقال با بیرون راندن یون های هیدروژن( H +)، از ماتریس مرکزی میتوکندری به فضای بین غشاهای خارجی و داخلی آن کوپل(جفت) می شود. این روند یک شیب پروتونی را ایجاد می کند که مولکول های ATP حامل انرژی را در میتوکندری توسط آنزیم ATP سنتاز تولید می نماید. اما اگر پروتون ها از خلال غشای داخلی به درون میتوکندری نشت کنند، این روند از این همزمانی و جفت بودن خارج می شود. این موضوع باعث ناهماهنگی در انتقال الکترون و خروج یون هیدروژن شده و از تولید کارآمد ATP می کاهد و درنتیجه باعث افزایش تقاضای داخل سلولی برای اکسیژن بمنظور تنفس بیشتر می گردد.
اختلال شدید در هماهنگی انتقال الکترون و خروج یون هیدروژن می تواند فیزیولوژی سلول را تغییر دهد و مهار این اختلال با ترکیبات مختلف باعث افزایش سطح اکسیژن سلولی، کاهش هیپوکسی و همچنین کاهش سطح پروتئین HIF-1α می شود. هر گونه دستکاری که منجر به کاهش فعالیت سلولی HIF-1α در بافت چربی شود، از نظر متابولیکی مفید است. بنابراین، درک بهتر چگونگی ایجاد اختلال در کوپل شدن انتقال الکترون و بیرون راندن یون هیدروژن(اختلال در ایجاد شیب پروتونی) و نحوه دستکاری آن می تواند به تغییر این وضعیت در جهت مطلوب کمک کند.
مطالعه ی قبلی این گروه نشان داده است که میزان مصرف اکسیژن در آدیپوسیت های سفید موش ها اگر این حیوانات با یک رژیم غذایی پر چرب تغذیه شوند، افزایش می یابد. محققان این فرضیه را مطرح کردند که افزایش میزان آزاد شدن اسید های چرب آزاد در خون حیوانات چاق، منجر به فعال شدن ANT2 می شود. فعالیت بیش از حد ANT2 منجر به افزایش نشت پروتونها به داخل میتوکندری می شود و سطح تنفس میتوکندریایی کوپل نشده را افزایش می دهد.
دکترSeo و همکارانش، مدلی از موشها را تولید کردند که در آنها بیان ژن Ant2 را به طور اختصاصی تنها در آدپیوسیت ها کاهش دادند، این کار به محققان این فرصت را می دهد که اثباتی برای این مکانیزم ارائه دهند. اولا، نویسندگان نشان دادند که تغذیه ی موشهای جهش یافته با رژیم غذایی پر چرب به اندازه ی موش های طبیعی، سبب چاقی این موشها می شود و هیچ گونه تفاوتی در وزن کلی بدن و فعالیت بدنی بین دو گروه وجود ندارد. با این حال، افزایش اندازه ی آدیپوسیتها (هایپرتروفی) موجب افزایش وزن بافت چربی در موشهای موتاسیونی Ant2 نسبت به گروه شاهد شده است، علیرغم ارتباط بین هایپرتروفی آدیپوسیتها و هیپوکسی که در مطالعات قبلی به آن اشاره شده است، نویسندگان دریافتند که فشار اکسیژن داخل سلولی[1] - غلظت اکسیژن در داخل سلول، که در هیپوکسی کاهش می یابد - در موش های حامل ژن Ant2 جهش یافته، بیشتر از گروه شاهد است. محققان نشان دادند که سلول برای حفظ غلظت اکسیژن داخلی خود، مصرف اکسیژن را کاهش می دهد، پس هیپوکسی به تغییرات در میزان عرضه ی اکسیژن یا دانسیته ی اکسیژن در عروق خونی مرتبط نیست، و این نشان می دهد که ANT2 عامل مهمی در تعیین میزان مصرف اکسیژن توسط آدیپوسیت ها در حیوانات چاق است.
بهبود غلظت اکسیژن در آديپوسیتهای موشهای Ant2-mutant مستقل از عملکرد ATP سنتاز بود، این موضوع نشان می دهد که با تغییرات در تنفس میتوكندریایی مرتبط نیست. در عوض، دکتر Seo و همکارانش تأیید کردند که این جهش باعث کاهش نشت پروتون ها از خلال غشای داخلی میتوکندری می شود که پتانسیل الکتریکی را در غشا افزایش می دهد و این، به نوبه خود، باعث افزایش کارایی تولید انرژی و کاهش اختلال در کوپلینگ انتقال الکترون و خروج یون هیدروژن (تصویر 1) شده و به همین ترتیب، بقای آدیپوسیت را نیز بهبود می بخشد.
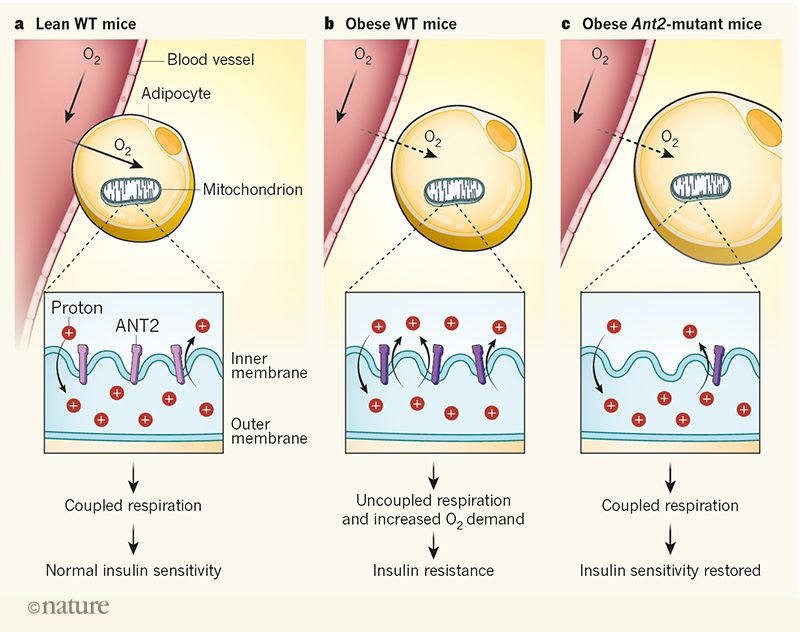
شکل 1 | آنزیم ANT2 در چاقی- a ، سلول های چربی (آديپوسیت ها) اکسیژن را از عروق خونی اطراف برای تنفس سلولی دریافت می کنند که در آن پروتون ها (یون های هیدروژن مثبت) از مرکز میتوکندری به فضای بین غشاهای داخلی و بیرونی منتقل می شوند، این کار باعث تولید پتانسیل غشایی می گردد که تولید انرژی را سبب می شود. آنزیم ANT2 باعث نشت پروتون ها از خلال غشای داخلی به درون میتوکندری می شود. این امر می تواند منجر به اختلال در کوپل شدن انتقال الکترون با راندن پروتون ها به فضای بین دو غشا شده و کارآیی تنفس را کاهش دهد، اما فعالیت ANT2 در موش های لاغر وحشی (WT) کم است. تنفس سلولی طبیعی، باعث تضمین حساسیت طبیعی بافت چربی به انسولین در این حیوانات می شود. b، در موش های چاق وحشی (WT)، آديپوسيت ها بزرگتر می شوند و اکسیژن کمتری(فلش های نقطه چین) دریافت می کنند که به دلیل عدم تامین خون کافی اتفاق می افتد. Seo و همکارانش گزارش می دهند که علاوه بر این، نیاز به اکسیژن نیز افزایش می یابد، زیرا فعالیت ANT2 در حیوانات چاق (با رنگ تیره نشان داده شده) افزایش می یابد و با کاهش پتانسیل غشا موجب اختلال در تنفس سلولی می شود. این منجر به مقاومت به انسولین (نشانه ی دیابت) در بافتهای اطراف آن می شود. c، کاهش بیان ژن Ant2 در آدیپوسیت موش های چاق باعث کاهش سطح ANT2، کاهش میزان اختلال در تنفس سلولی و در نتیجه کاهش نیاز به اکسیژن می گردد و بنابراین حساسیت به انسولین را بهبود می بخشد. به طرز شگفت آوری، آدیپوسیتهای این حیوانات جهش یافته، بزرگتر از موشهای چاق وحشی هستند، اما حساسیت بالاتری را به انسولین نشان می دهند.
طیف وسیعی از سلول های ایمنی در هنگام رشد بافت چربی وارد عمل می شوند که موجب پاسخ های التهابی و اسکارهای بافتی به نام فیبروز می شوند. اما Seo و همکارانش نشان دادند که بهبود عملکرد میتوکندری ناشی از کاهش فعالیت Ant2بطور خاص در آدیپوسیتها این پاسخ را کاهش می دهد. همچنین اگر از طریق مهندسی ژنتیک چگالی عروق را در بافت چربی افزایش دهیم، این پاسخ بهبود می یابد. همانطور که انتظار می رود این کاهش در التهاب و فیبروز منجر به بهبود تحمل گلوکز و افزایش حساسیت به انسولین در کبد موشهای حامل ژن جهش یافته ی Ant2 می شود. علاوه بر این، محققان نشان دادند که حذف Ant2 در آدپیوسیت موش هایی که در حال حاضر به عدم تحمل گلوکز و مقاومت به انسولین مبتلا هستند، می تواند این اثرات را معکوس کند.
این یافته ها به دلایل مختلف مورد توجه هستند. اول این که، در بسیاری از مطالعات بر نیاز به تشکیل عروق کافی در بافت چربی برای جلوگیری از هیپوکسی تاکید شده است، اماSeo و همکارانش آديپوسیت را به عنوان یک نیروی محرکه برای هیپوکسی مطرح کردند، و نشان دادند که نقص در سلول چربی منجر به کاهش میزان اکسیژن داخل سلولی می شود، و می تواند تغییرات متابولیکی گسترده تری را ایجاد کند. دوم این که، در وزن آديپوسیتهای موش های حامل ژن Ant2 جهش یافته، یک افزایش وزن کلی مشاهده می شود. این یافته، مخالف تصور همیشگی است، زیرا هایپرتروفی آدیپوسیتها به طور کلی با اختلال در متابولیسم ارتباط دارد، بنابراین این مشاهدات نیازمند بررسی بیشتر هستند. یک توضیح احتمالی این است که سطح HIF-1α در سلولهای هایپرتروفی کاهش مییابد و باعث افزایش بقای آنها می شود. سوم این که، محققان نشان دادند که غلظت اکسیژن درون سلولی در بافت چربی افراد چاق که از نظر متابولیکی طبیعی هستند بیشتر از افراد چاق با متابولیک غیر طبیعی است. این موضوع سازگار با حساسیت به انسولین در این افراد است، که نشان می دهد یافته های نویسندگان ممکن است دارای ارتباط بالینی باشند.
تحقیق اخیر دکترSeo و همکارانش، مدولاسیون ANT2 را به عنوان یک استراتژی بالقوه برای بهبود نقایص متابولیکی سیستمیک، از جمله دیابت نوع 2، تعریف می کند. در کنار این واقعیت که ANT2 قبلا بعنوان یک هدف ضد سرطان پیشنهاد شده است، این امر باعث می شود که مدولاتورهایANT2 ، نامزدهای اصلی برای توسعه ی داروهای جدید باشند. این امر با توجه به این واقعیت که دکتر Seo و همکارانش تنها تا اندازه ای بیان Ant2 را در مطالعه ی فعلی مهار کردند( نه حذف کامل ژن) بسیار مطلوب است. بنابراین، مولکولهای کوچک دارویی که بتوانند فعالیت آنزیم مورد نظر را تنها تا حدی مسدود کنند ممکن است اثرات مورد نظر را فراهم نمایند. در آینده تلاش ها بر شناسایی چنین مهار کننده هایی متمرکز خواهد شد.
منبع:
https://www.nature.com/articles/d41586-018-07248-6